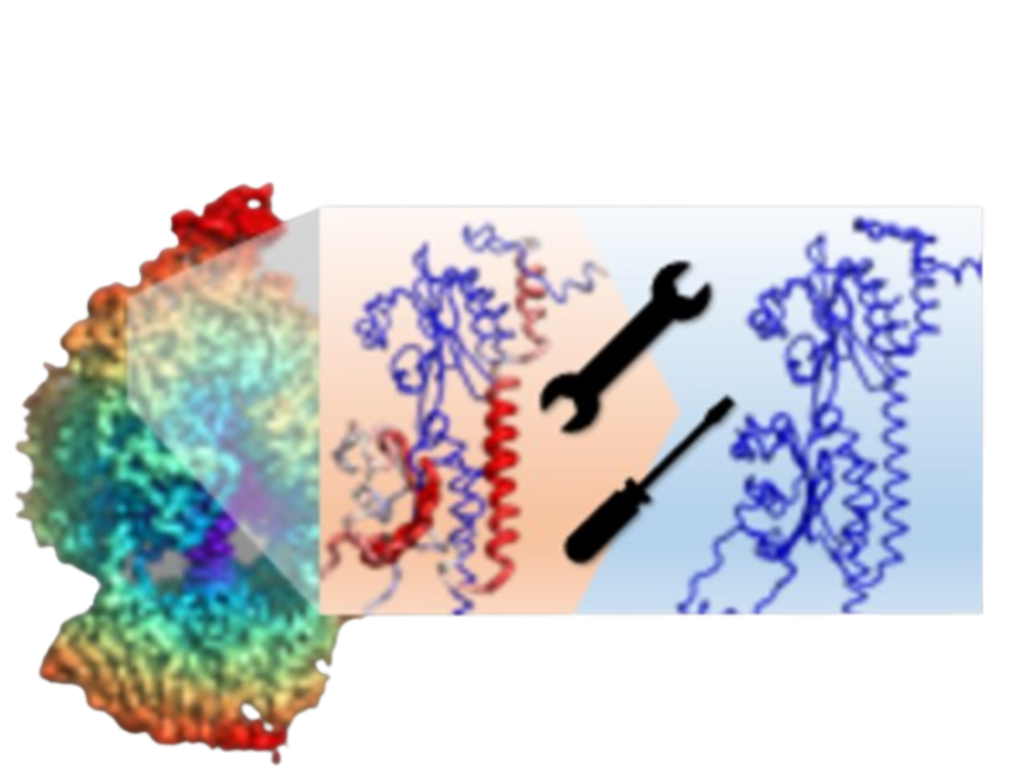
CryoZeta is a de novo macromolecular structure modeling tool that integrates cryo-EM density information with a diffusion-model-based structure prediction pipeline.
It jointly leverages sequence information and cryo-EM map features to generate atomic models consistent with experimental density, and is effective for proteins, protein-nucleic acid complexes, and nucleic acid-only systems at resolutions up to ~10 Å. CryoZeta supports complexes with a total sequence length of up to ~2,700 residues or bases. If the total length exceeds this threshold, the system automatically switches to "cycle mode." Please note that each individual chain must not exceed the ~2,700 length limit.
If encounter problems, please contact Daisuke Kihara (dkihara@purdue.edu)
Please simply click "Upload" when you filled all input fields.
Density Histogram (|ρ|)
0.00
Contour: 0.00
0.00
Please do not input 0, you must provide a contour to remove the outside very noisy regions
You can either upload a sequence file in FASTA format or paste the raw sequences directly into the sequence fields.
Please provide at least one sequence (Protein, DNA, or RNA).
0/500
This note will help you distinguish jobs later.