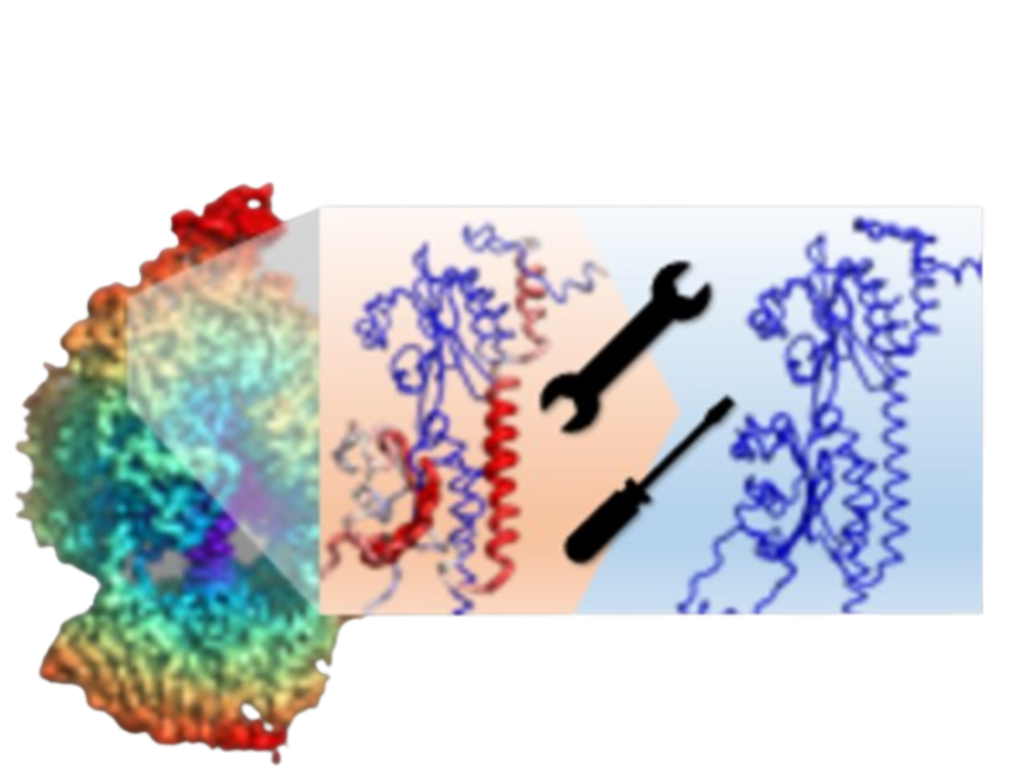
DiffModeler uses a diffusion model to automatically build full protein complex structures from cryo-EM maps at up to 20 Å resolution. This sequence version searches for template structures automatically.
Templates are searched by BLASTP against the PDB and AlphaFold Database.
Only hits with an E-value of 0 in the PDB95 search are considered. The first hit whose chain length falls within 90–110% of the expected length is selected. If no hit is found in PDB95, the search falls back to the combined PDB + AlphaFold Database.
If you only know some sequences for the cryo-EM map, you can still run DiffModeler and get good partial structures.
If you encounter problems, please contact Daisuke Kihara (dkihara@purdue.edu) or Han Zhu (zhu773@purdue.edu).
Example Input
Map File: 6824.mrcSequence: 6824.fastaContour level: 3.0
Resolution: 5.8
Example Result
View example job
Video Tutorial:
Please click "Upload" when you have filled in all input fields.
Density Histogram (|ρ|)
0.00
Contour: 0.00
0.00
Determine an appropriate contour level for fitting your protein structure. You can also find it interactively on the submission page after uploading your map.
The tool will not work properly without an appropriate contour level.
The map resolution is required as input. If the resolution is 2 Å or better (high resolution), the diffusion step will be skipped.
Paste sequences directly into the text fields on the submission page. You can also upload a FASTA file (see the tutorial for the format).
Select Yes for domain-based modeling. The server will use SWORD2 to split each chain into domains.
0/500
This note will help you distinguish jobs later.