DiffModeler Tutorial
Last updated on May 26, 2026
Video Tutorial
Overview
DiffModeler is a computational tool using a diffusion model to automatically build full protein complex structure from cryo-EM maps at 0-20 Å resolution.
Overall Pipeline
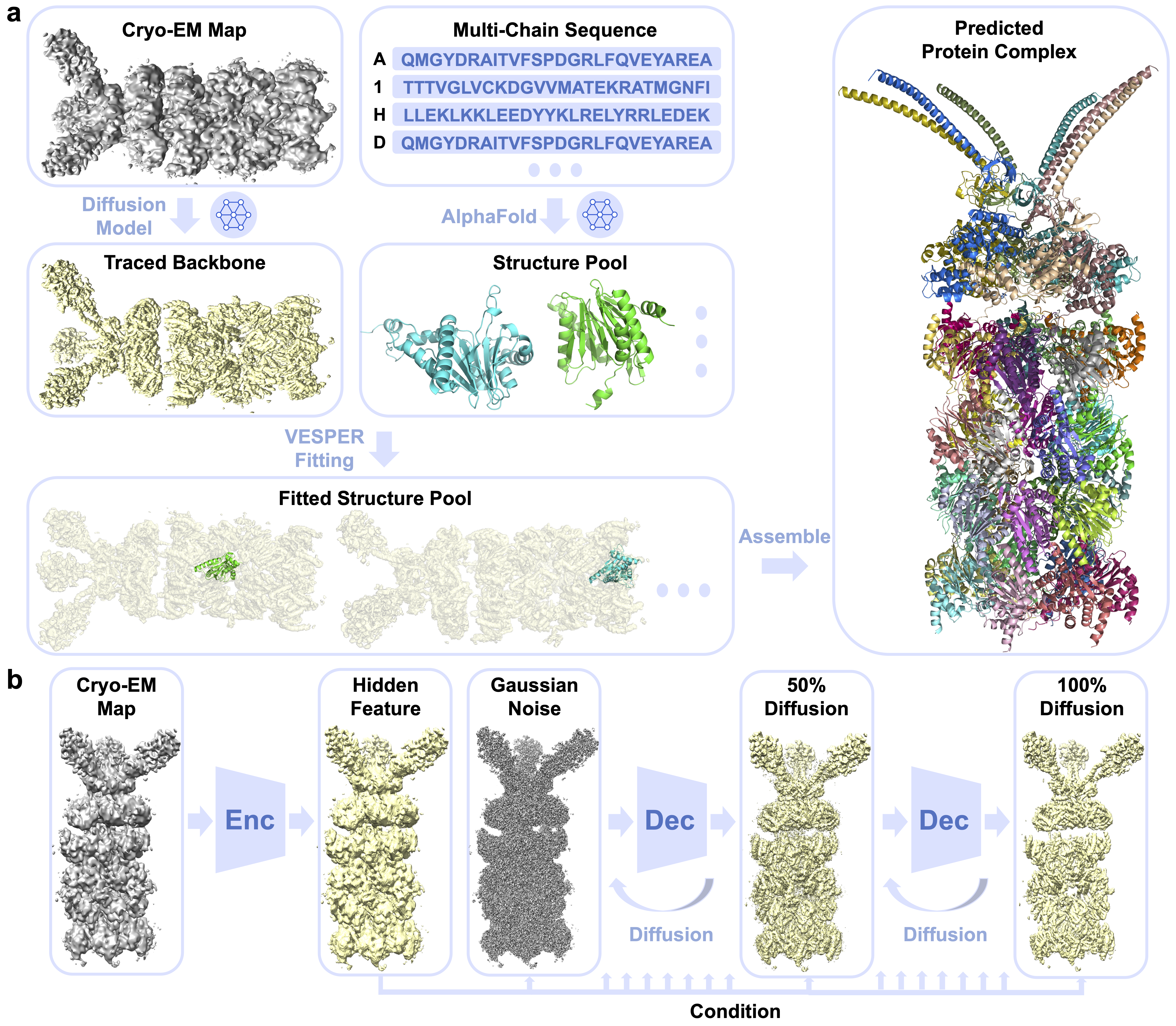
- Backbone tracing from cryo-EM maps at intermediate resolution via diffusion model.
- Single-chain structure prediction by AlphaFold.
- Single-chain structure fitting using VESPER.
- Protein complex modeling by assembling algorithms.
Input Files
DiffModeler:- 3D cryo-EM map in
.mrcor.mapformat - single chain pdb files in
.pdbformat
- 3D cryo-EM map in
.mrcor.mapformat - Sequence files in
.fastaformat
Output Files
Full protein complex structure in .cif file
Job Submission
- Prepare Input Map
Collect 3D cryo-EM map from microscope in
.mrcor.mapformat.
Example map
You can also find many maps in EMDataResource as testing examples.
- Prepare Input pdb files
- Prepare Input sequence file(Only if you are using DiffModeler(seq))
- Decide the contour level
You can choose and adjust the contour level interactively using the online visualizer on the job submission page after uploading your map. Otherwise, please make sure your contour level is lower than your focused region.
This is absolute density threshold, not standard deviation.
Please do not input 0, you must provide a contour to remove the outside very noisy regions. - Decide the resolution
- Submit your job
Once you collected the input files, please submit your job here (DiffModeler(seq)->here) For each input field, please input the files/info collected before.
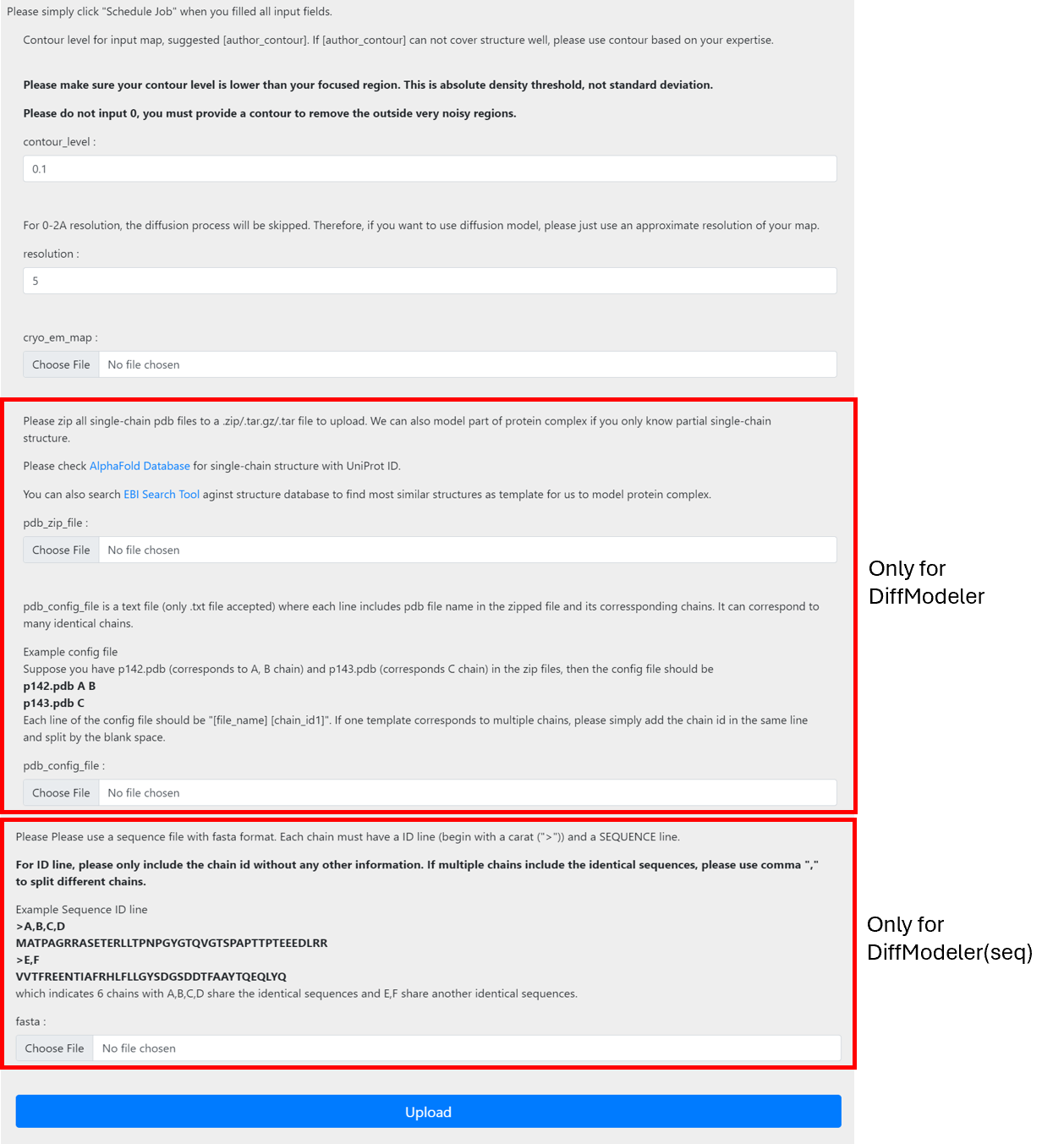
Once you finished input, simply click the upload button to submit jobs. After submission, you will be redirected to the View Job page. If you are not registered, please bookmark the link. Once the job is done, you can view jobs from this link. If you are registered, you will receive email notifications once job is done and you can also check job status from my jobs list under job manager.
- View your job results
Once job is done, you can check the modeled structure from the link bookmarked before. Here you can also download the modeled structure in
.pdbformat by clicking the “Download Outputs” button. You can also visualize the 3D cryo-EM map online to check its consistency with the modeled structure. For more detailed instructions, please see the Instructions in the same page.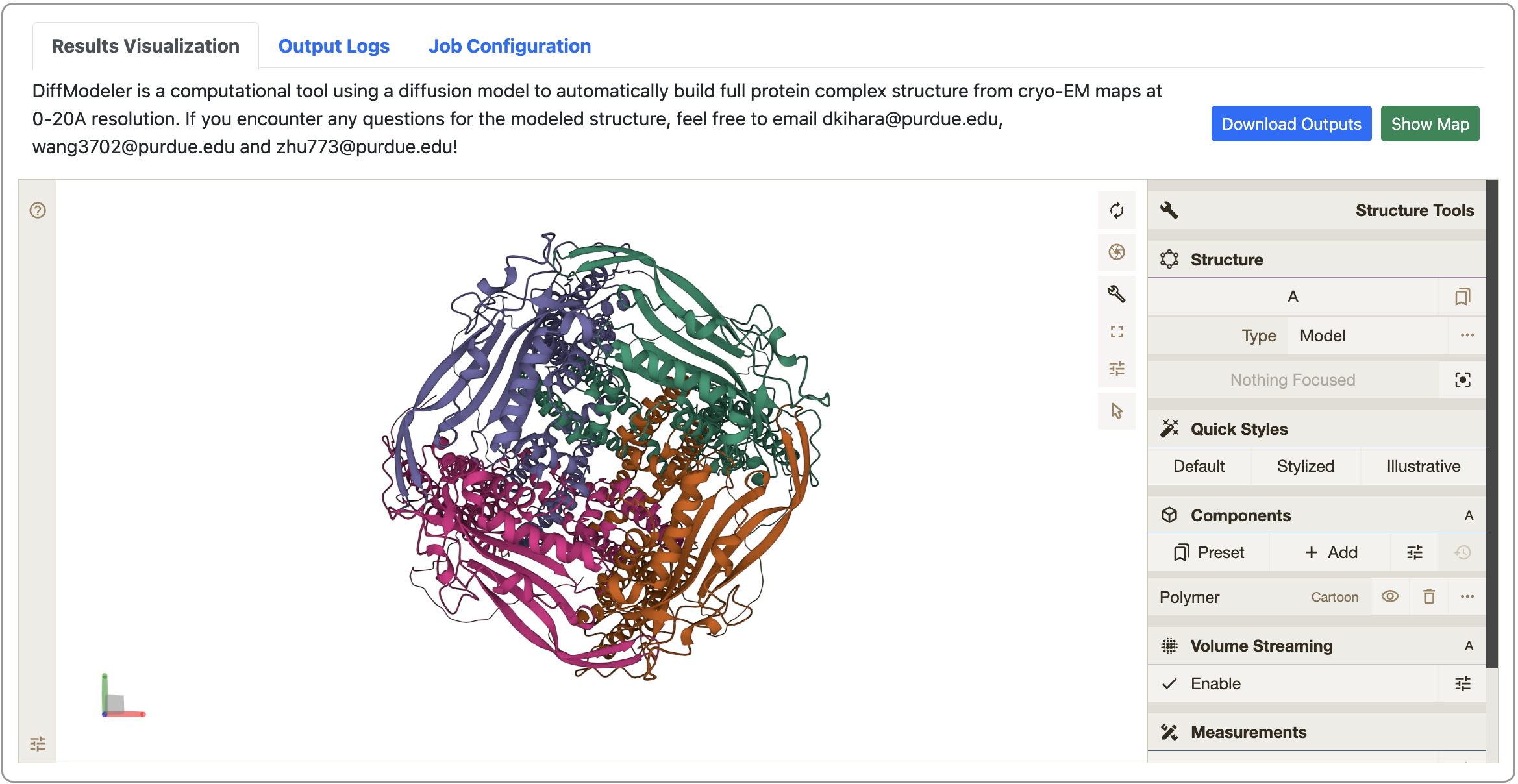
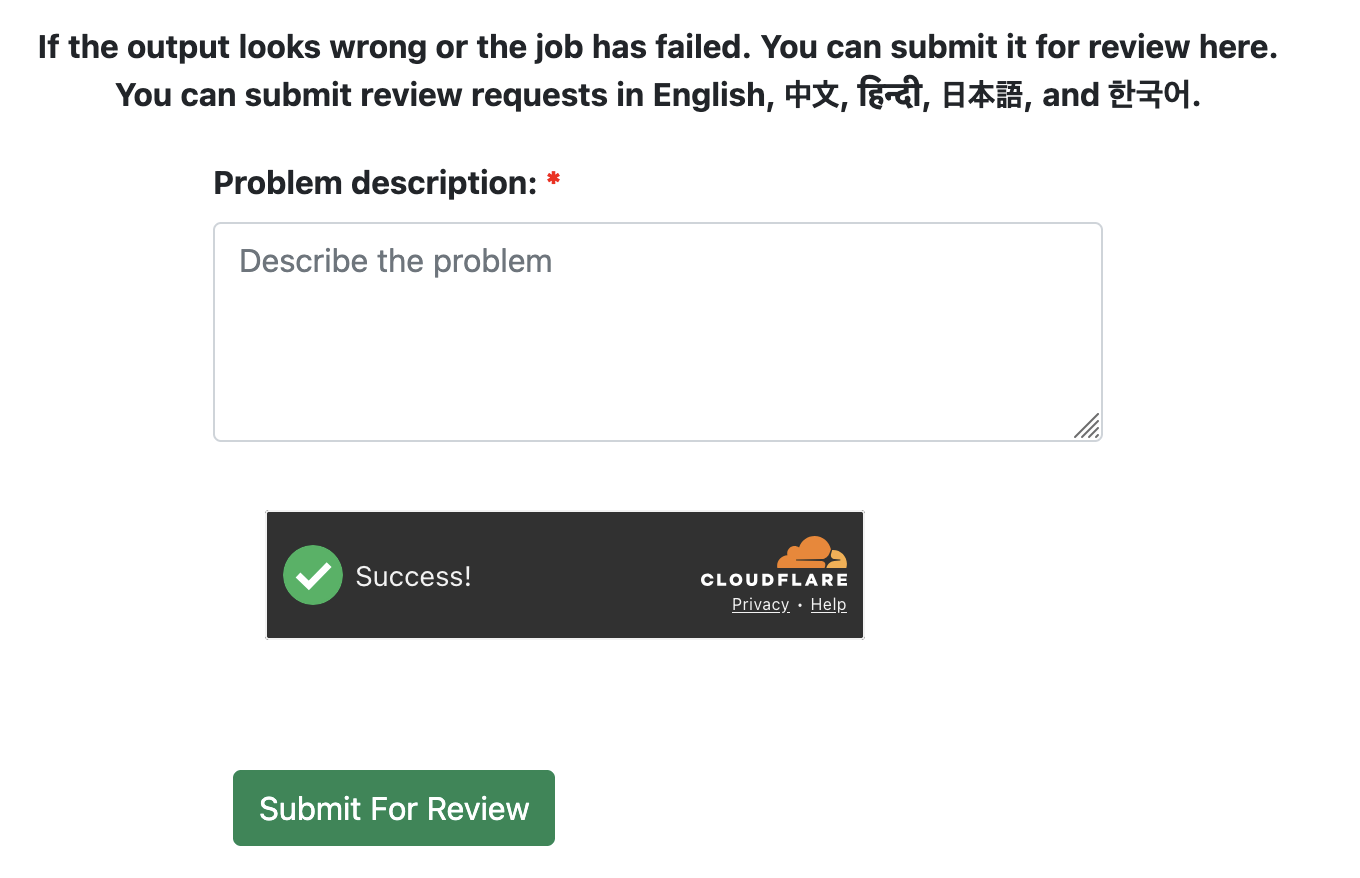
- Submit for backend review (optional)
If you noticed any strange outputs or job failure on your side, please submit a backend review by using the field at the bottom of the View Job page. We will get back to you as soon as possible.
Please upload single-chain pdb files with number of copies.
e.g. If you have a protein A.pdb and want to include the same chain twice, please write 2 in the number section.
We can also model part of protein complex if you only know partial single-chain structure.
Please check AlphaFold Database for single-chain structure with UniProt ID.
You can also search EBI Search Tool aginst structure database to find most similar structures as template for us to model protein complex.
Please use a sequence file with fasta format or enter/type it directly in the text area on the submission page. Each chain must have a ID line (begin with a carat (">"))
and a SEQUENCE line.
For ID line, please only include the chain id without any other information. If multiple chains include the
identical sequences, please use comma "," to split different chains.
Example Sequence ID line:
>A,B,C,D
MATPAGRRASETERLLTPNPGYGTQVGTSPAPTTPTEEEDLRR
>E,F
VVTFREENTIAFRHLFLLGYSDGSDDTFAAYTQEQLYQ
which indicates 6 chains with A,B,C,D share the identical sequences and E,F share another identical sequences.
For 0-2A resolution, the diffusion process will be skipped.
Therefore, if you want to use diffusion model, please just use an approximate resolution of your map.
Availability (Other)
- GitHub
Full code is available here.
Reference
Wang, X., Zhu, H., Terashi, G., Taluja, M., & Kihara, D. (2024). DiffModeler: Large macromolecular structure modeling for cryo-EM maps using a diffusion model. Nature Methods. https://doi.org/10.1038/s41592-024-02479-0