DAQ-Refine Tutorial
Overview
DAQ-refine is a protocol using DAQ score to evaluate protein models from cryo-EM maps (up to 5 A resolution) and employs a modified AlphaFold2 to refine regions with potential errors.
* Drag to the right: Refined model.
* Drag to the left: Original model.
- If the image does not fit in the box, please reload the page.
Overall Pipeline
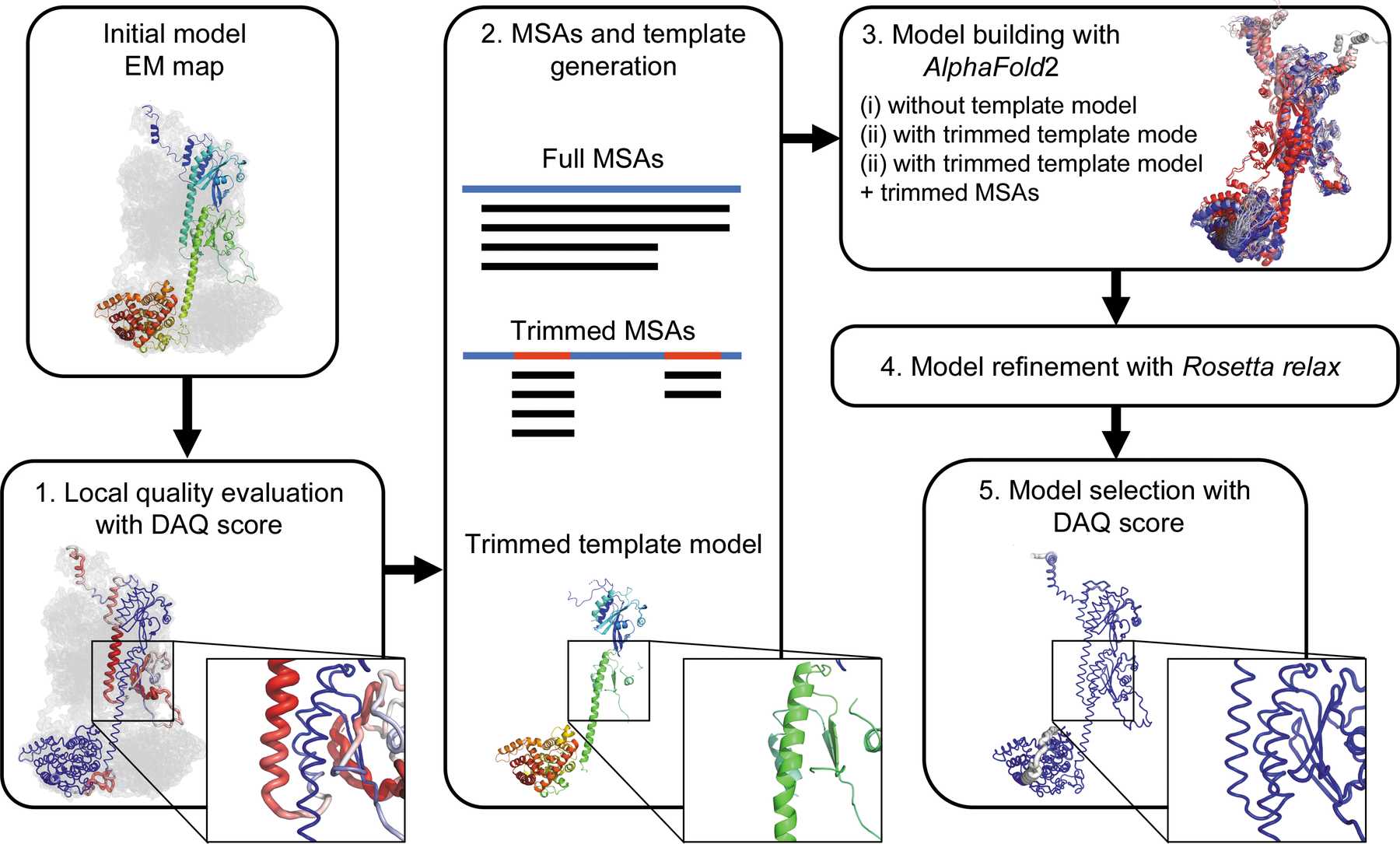
- Initial model evaluation with the DAQ score. The DAQ(AA) scores along the model range with a color scale from red [DAQ(AA) < −1.0] to blue [DAQ(AA)> 1.0]
- MSA and template-model generation. Full MSAs are computed by MMseqs2 in ColabFold. Trimmed MSAs are generated by masking alignment data corresponding to positions in the full MSAs where the DAQ(AA) score is positive. The trimmed template model is generated by removing residue positions where the DAQ(AA) score is negative or zero from the initial model.
- Model building by AlphaFold2. Three strategies (AF2 with full MSAs, AF2 with full MSAs + trimmed template model and AF2 with trimmed MSAs + trimmed template model) are performed.
- Models are refined with Rosetta relax in the EM map.
- Finally, the top-ranked model by DAQ(AA) score is selected as the final model.
Input Files
- Protein Structure in .pdb format
- 3D cryo-EM map in .mrc format
- Sequence files in .fasta format
Output Files
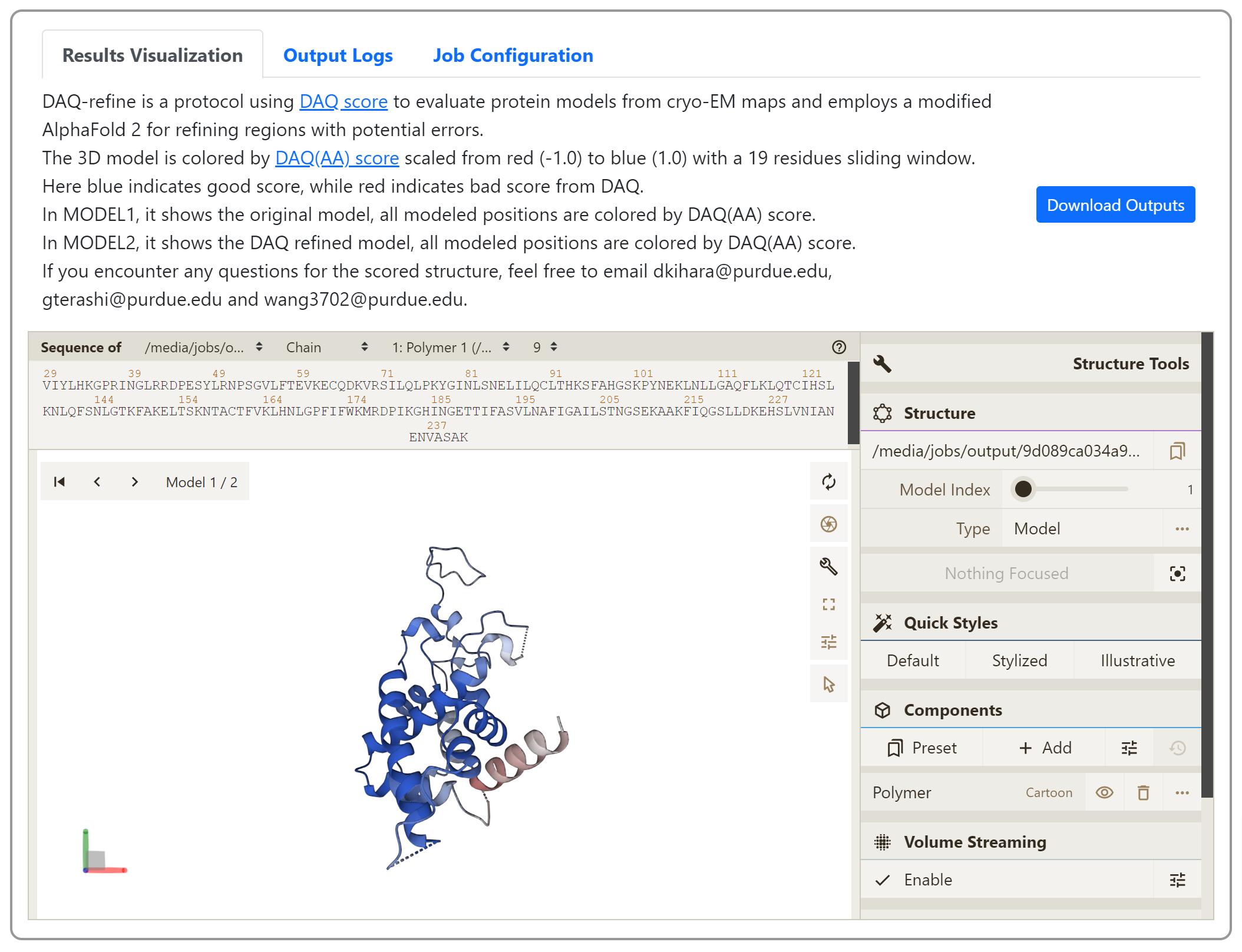
3D model produced after refining regions with potential errors. The model is colored by DAQ(AA) score scaled from red (-1.0) to blue (1.0) with a 19 residues sliding window.
Job Submission
- Prepare Input Map
Collect 3D cryo-EM map from microscope in .mrc and .map format.
Example map
You can also find many maps in EMDataResource as testing examples.
- Protein Structure
Upload your structure model in pdb format
Example pdb
- Sequence File
- Submit your job
Once you collected the input files, please submit your job here. For each input field, please input the files/info collected before.
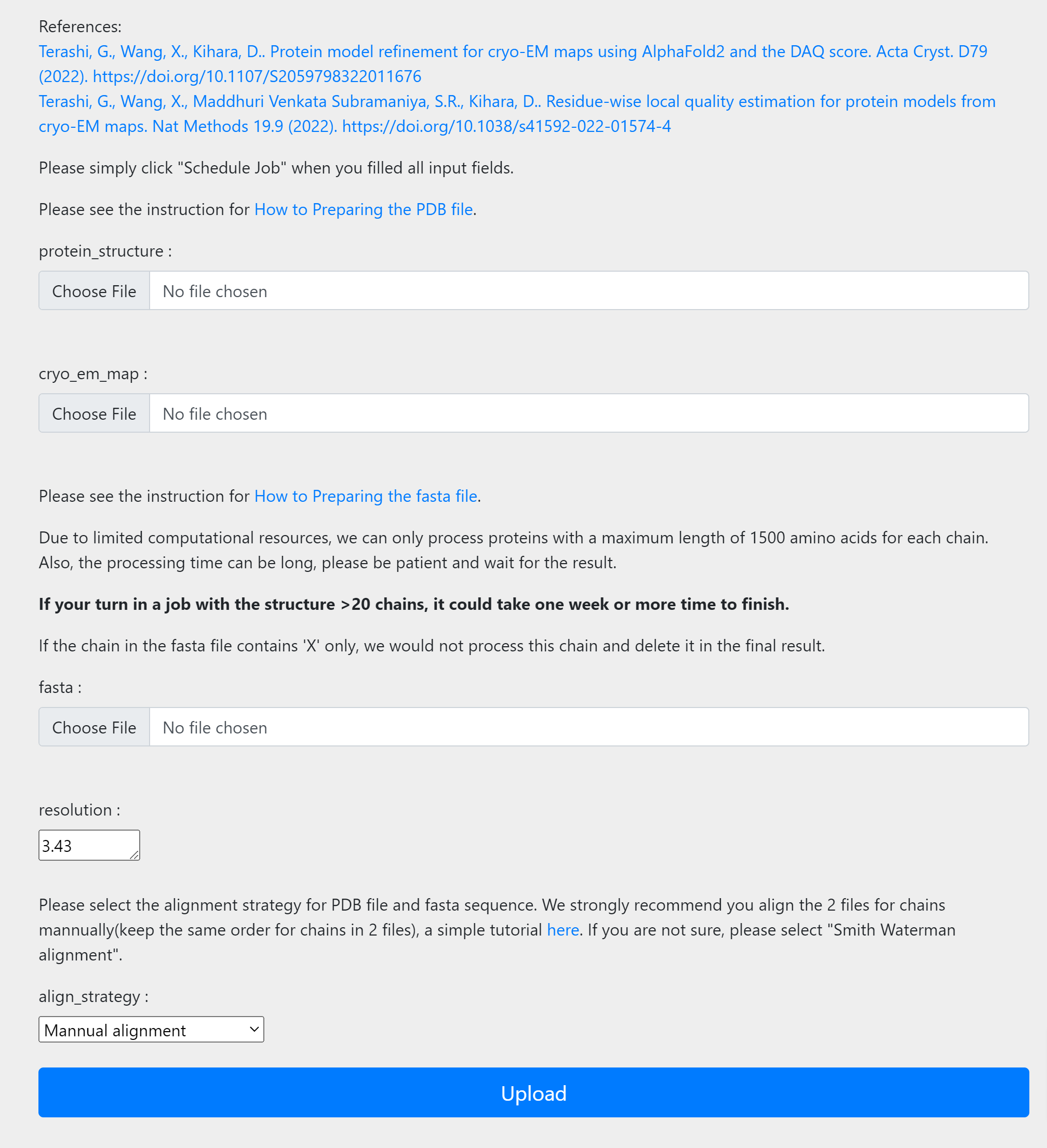
Once you finished input, simply click the upload button to submit jobs. After submission, you will be redirected to the “view job“ page. If you are not registered, please bookmark the link. Once the job is done, you can view jobs from this link. If you are registered, you will receive email notifications once job is done and you can also check job status from my jobs list under job manager.
- View your job results
Once job is done, you can check the modeled structure from the link bookmarked before. Here you can also download the modeled structure in .pdb format by clicking the “Download Output” button. You can also visualize the 3D cryo-EM map online to check its consistency with the modeled structure. For more detailed instructions, please see the “Instructions” in the same page.
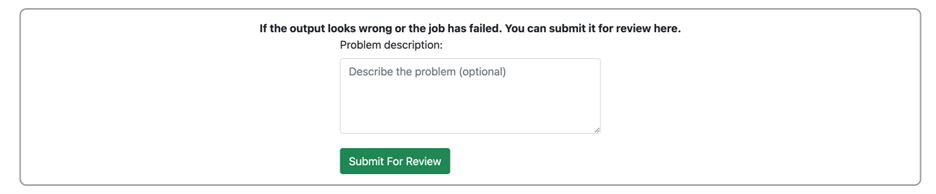
- Submit for backend review (optional)
If you noticed any strange outputs or job failure on your side, please submit a backend review by using the field in the bottom of the “View Job” page. We will get back to you as soon as possible.
Availability (Other)
- GitHub
This github contains a modified ColabFold notebook and our tools.
- Google
Colab
Step-by-step instructions are available.
Reference
Genki Terashi, Xiao Wang, Daisuke Kihara. Protein model refinement for cryo-EM maps using AlphaFold2 and the DAQ score. Acta Crystallographica Section D Structural Biology, 79, 10–21, (2023). https://doi.org/10.1107/s2059798322011676